Accelerating molecular dynamics simulations with MPI and GPU
In this article, we will explore techniques for speeding up molecular dynamics simulations with the help of HPC. We'll be focusing on utilizing NAMD as our simulation software and examining the ApoA1 in a water box environment.
Molecular dynamics (MD) simulation
Molecular dynamics simulation is a computational technique used to study the movements and interactions of atoms and molecules over time. By simulating the behavior of these particles, scientists can gain insights into various phenomena, such as protein folding, chemical reactions, and material properties.
Molecular dynamics simulations are computationally intensive and require significant computational resources. This makes them an excellent candidate for acceleration using High-Performance Computing (HPC). By harnessing the power of HPC, researchers can significantly reduce simulation time and tackle more complex and realistic systems. This acceleration allows for faster data analysis, improved understanding of molecular behavior, and enables scientists to make discoveries more efficiently.
NAMD
NAMD is a widely used software program for molecular dynamics (MD) simulations. It stands for "Nanoscale Molecular Dynamics" and is designed to study large biological systems' behavior and movements, such as proteins, nucleic acids, and lipid membranes.
NAMD utilizes parallel computing techniques to accelerate MD simulations and can run on high-performance computing (HPC) systems. It provides extensive features and algorithms for accurately modeling complex molecular systems, including force fields, energy minimization, and simulation analysis tools.
NAMD is highly regarded for its scalability, efficiency, and ability to handle large biomolecular systems. It has become a valuable tool for researchers in biochemistry, biophysics, and drug discovery, enabling them to gain insights into the dynamics and interactions of biological molecules at the atomic level.
Parallel computing
Parallel computing refers to the use of multiple processors or computing resources to perform computations simultaneously. It involves breaking down a complex problem into smaller tasks that can be executed concurrently, allowing faster and more efficient computation.
Parallel computing is crucial to High-Performance Computing (HPC) because it enables the simultaneous execution of multiple tasks, leading to significant improvements in computational speed and performance. HPC systems, with their large-scale infrastructure and interconnected computing nodes, are designed to leverage parallel computing techniques.
Parallel computing can be used for MD simulation to significantly speed up the computational process. MD simulations involve simulating the movements and interactions of a large number of atoms or molecules over time, which requires performing numerous calculations simultaneously. By dividing the workload among multiple processors or computing nodes in parallel, parallel computing allows for faster execution of these calculations. This speeds up the overall simulation time and enables researchers to study more complex systems or perform longer simulations that would otherwise be impractical with sequential computing.
ApoA1
The ApoA1 example case is a widely used benchmark simulation for the NAMD molecular dynamics software package. It is a simulation of an Apolipoprotein A1 (ApoA1) molecule in a water box, and it is often used to test the performance and accuracy of NAMD on large-scale molecular dynamics simulations.
Apolipoprotein A1 (ApoA1) is a protein that plays a critical role in transporting cholesterol and other lipids in the bloodstream. It is the major protein component of high-density lipoprotein (HDL), also known as "good cholesterol," which helps to remove excess cholesterol from the blood vessels and transport it to the liver for processing and excretion.
The structure of ApoA1 is complex and consists of multiple domains that interact with lipids, other proteins, and cellular receptors. It is a large protein with a molecular weight of approximately 28 kDa and a length of 243 amino acids, and it undergoes significant conformational changes depending on its interactions with other molecules.
Setup the environment
We will start by downloading the environment files from the NVIDIA ngc-examples Git Hub repository.
cd $HOME
wget -O - https://gitlab.com/NVHPC/ngc-examples/raw/master/namd/3.0/get_apoa1.sh | bash
# apply the following 2 modifications to the environment to:
# 1) lengthen the simulation to have a clearer comparison to the performance
# 2) output a dcd file for visualizing the protein movement later
vi ~/apoa1/apoa1.namd
32c32
< numsteps 500
---
> numsteps 1000
35c35,38
< outputname /usr/tmp/apoa1-out
---
> set output output
> outputname $output
> dcdfile ${output}.dcd
> dcdfreq 10Run with one CPU core
# go to the environment directory
cd ~/apoa1/
# load the NAMD environment module
module load GCC OpenMPI NAMD
# start the simulation with 1 CPU core
srun -p batch namd2 apoa1.namd
Charm++> Running on MPI version: 3.1
Charm++> level of thread support used: MPI_THREAD_SINGLE (desired: MPI_THREAD_SINGLE)
Charm++> Running in non-SMP mode: 1 processes (PEs)
Charm++> Using recursive bisection (scheme 3) for topology aware partitions
...
TIMING: 100 CPU: 47.0369, 0.457802/step
...Multicores
# run the simulation with 16 cores
sbatch -p batch -n16 -N1 -o namd.out --wrap "charmrun +p16 namd2 apoa1.namd"
# inspect the live output
tail -f namd.out
...
TIMING: 100 CPU: 3.12054, 0.0302191/step
...Multinodes
# run the simulation with 32 cores on 2 nodes
sbatch -p batch -n32 -N2 -o namd.out --wrap "charmrun +p32 namd2 apoa1.namd"
# inspect the live output
tail -f namd.out
...
TIMING: 100 CPU: 2.09707, 0.0194772/step
...One GPU
export CONT="/pfss/containers/ngc.namd.3.0-beta2.sif"
# run the simulation with 1 1g.10gb A100 MIG instance (~1/7 computing power of A100)
sbatch -p gpu -c4 --gpus 1g.10gb:1 -o namd.out --wrap "
singularity exec --nv $CONT namd3 +p1 +devices 0 +setcpuaffinity apoa1.namd
"
# inspect the live output
tail -f namd.out
...
TIMING: 100 CPU: 1.588, 0.0151/step
...Multiple GPUs
# run the simulation with 4x 1g.10gb A100 MIG instance (~4/7 computing power of A100)
sbatch -p gpu -n4 -N1 -c4 --gpus 1g.10gb:4 -o namd.out --wrap "
singularity exec --nv $CONT namd3 +p4 +devices 0 apoa1.namd
"
# inspect the live output
tail -f namd.out
...
TIMING: 100 CPU: 0.967313, 0.0092312/step
...Result
| Time used for simulating 1,000 time steps |
|
| 1 CPU core |
490s |
| 16 cores / one node |
36s |
| 32 cores / 2 nodes |
23s |
| 1x 1g.10gb |
16.3s |
| 4x 1g.10gb |
8.9 |
Analyze output with VMD over VNC
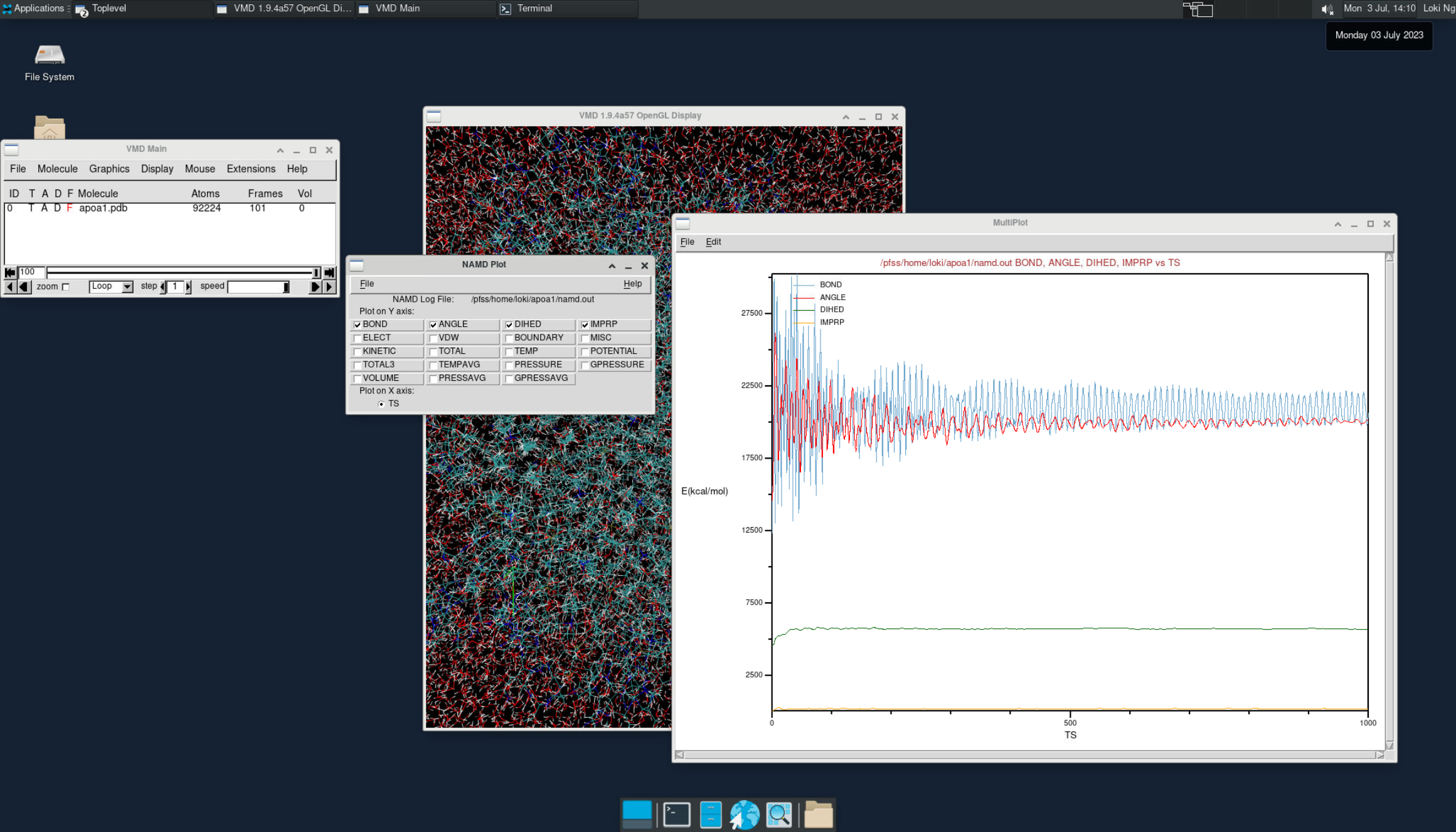
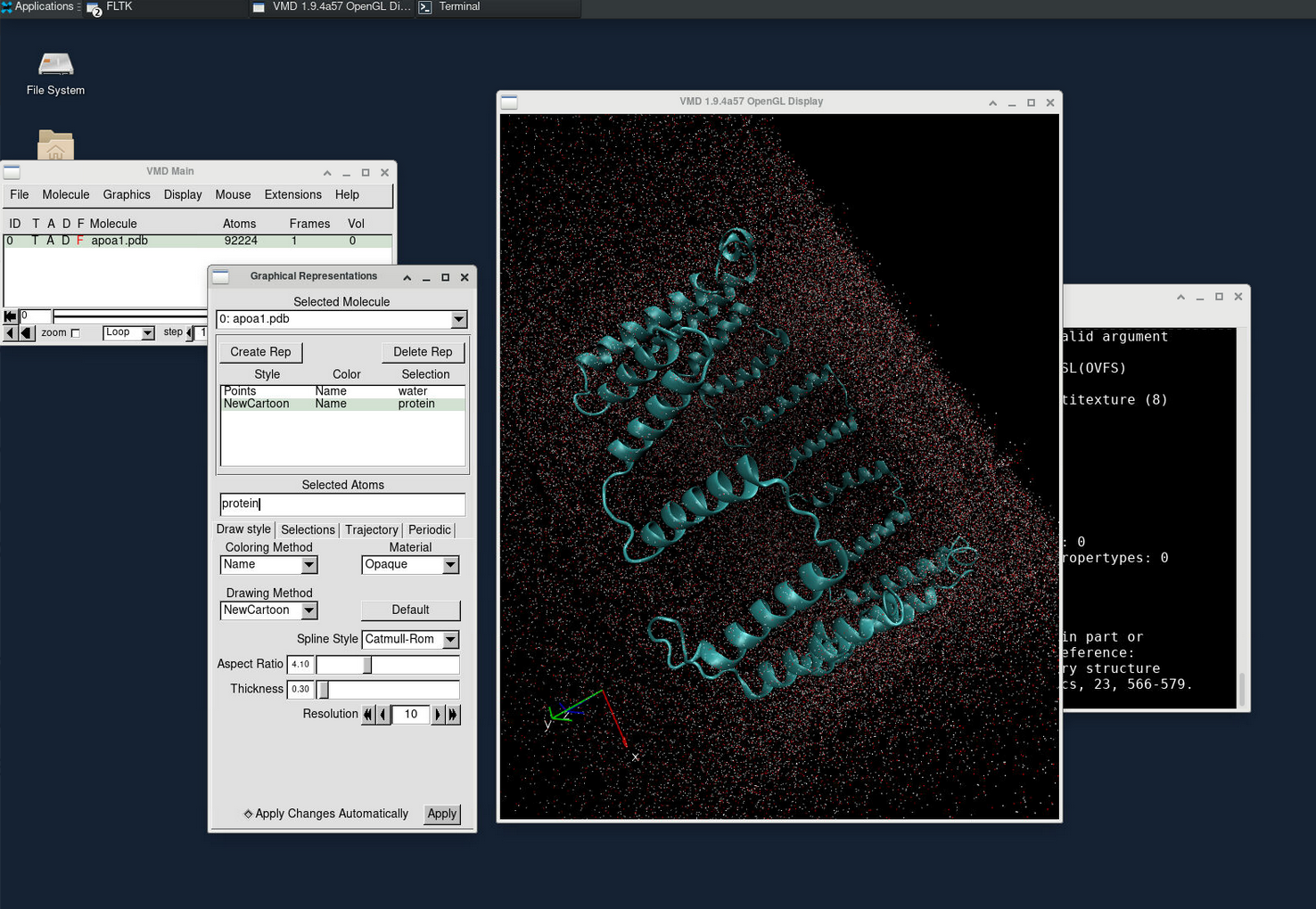
VMD is commonly used for visualizing and analyzing the output of NAMD simulations. As a standalone desktop application, we can request a VNC session on a compute node to utilize VMD.
Click the VNC link in the running jobs viewer to connect to our web VNC client.
Inside the VNC session, open a new terminal and input the following statement to open VMD.
module load GCC OpenMPI VMD
vmdTo view the simulation results in a 3D animation, create a new molecule and load the apoa1.pdb and output.dcd files. This will allow you to visualize the molecular dynamics.
Additionally, you can plot various energy levels over time by going to Extension > Analysis > NAMD plot. This feature lets you analyze and track different energy trends during the simulation.