Accelerate FASTQ to BAM conversion using GPU and Parabricks
Refs to Parabricks: fq2bam (FQ2BAM + BWA-MEM)
Parabricks fq2bam is a software tool that can generate BAM/CRAM output from one or more pairs of FASTQ files. This tool takes advantage of the parallel computing capabilities of GPUs to speed up the analysis process.
To use Parabricks, users can provide input files in FASTQ format and specify the reference genome they wish to use for alignment. The software uses a proprietary algorithm to perform read alignment, variant calling, and quality control. The output is then generated in BAM or CRAM format, depending on the user's preference.
In this case study, we will align our sample and reference genome for further comparison and analysis. Following is our reference genome.
Reference Genome: human_g1k_v37.fasta
Sample Data Source: SRA SRR7733443
Number Of Read: 2 x 5M bp
Read length: 150bp
We followed the steps below to collect the performance results:
First, we need to use the scratch directory. Please prepare about 50GB of free space for this experiment.
We have prepared samples (1M, 5M, 110M) and their index in OAsis. You may skip steps 1 to 4 by utilizing them.
To utilize the prepared data, run: cp /pfss/toolkit/parabricks_data $SCRATCH/parabracks
- Download the sample genome for analysis.
- Download the SRA toolkit from https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software#header-global
- tar xfzv sratoolkit.2.10.5-centos_linux64.tar.gz
- sratoolkit.2.10.5-centos_linux64/bin/fastq-dump -X 5000000 --split-files SRR9932168
- Download the reference genome.
- Download reference from http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/human_g1k_v37.fasta.gz
- Download reference from http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/reference/human_g1k_v37.fasta.gz
- Index the genome.
- Leverage our Parabricks container, which already includes samtools.
-
singularity run --nv /pfss/containers/clara-parabricks.4.0.0-1.sif bash samtools faidx /human_g1k_v37.fasta - install bwa and use it to index human_g1k_v37.fasta
-
# install bwa git clone https://github.com/lh3/bwa.git cd bwa make # request a compute node to perform the indexing srun --pty -p batch --cpus-per-task=32 --mem=100G bash # in the prompt, run: ./bwa index ../human_g1k_v37.fasta
-
- Leverage our Parabricks container, which already includes samtools.
- Download Known Site
- download 00-common_all.vcf from https://ftp.ncbi.nih.gov/snp/organisms/human_9606_b150_GRCh37p13/VCF/
- singularity run /pfss/containers/gatk.sif gatk IndexFeatureFile -I /00-common_all.vcf
- Run Parabricks
- We utilize one NVIDIA A100 here, be careful that Parabricks currently does not support MIG.
-
srun -p gpu -c 16 --gres=gpu:a100:1 --mem=128g --pty bash singularity run --nv /pfss/containers/clara-parabricks.4.1.0-1.sif bash cd $SCRATCH/parabricks mkdir tmp NVIDIA_VISIBLE_DEVICES="0" pbrun fq2bam \ --num-gpus 1 \ --ref ./human_g1k_v37.fasta \ --in-fq ./SRR9932168_1.fastq ./SRR9932168_2.fastq \ --out-bam ./mark_dups_gpu.bam \ --tmp-dir ./tmp \ --knownSites ./00-common_all.vcf \ --out-recal-file ./recal_gpu.txt
- Result
- The output should be something like this:
-
[main] CMD: /usr/local/parabricks/binaries//bin/bwa mem -Z ./pbOpts.txt -F 0 /pfss/scratch02/appcara/parabricks_2/human_g1k_v37.fasta /pfss/scratch02/appcara/parabricks_2/5M_sample/SRR9932168_1.fastq /pfss/scratch02/appcara/parabricks_2/5M_sample/SRR9932168_2.fastq @RG\tID:SRR9932168.1.1\tLB:lib1\tPL:bar\tSM:sample\tPU:SRR9932168.1.1
[main] Real time: 73.634 sec; CPU: 936.517 sec
[PB Info 2023-Sep-13 17:21:43] ------------------------------------------------------------------------------
[PB Info 2023-Sep-13 17:21:43] || Program: GPU-BWA mem, Sorting Phase-I ||
[PB Info 2023-Sep-13 17:21:43] || Version: 4.1.0-1 ||
[PB Info 2023-Sep-13 17:21:43] || Start Time: Wed Sep 13 17:20:29 2023 ||
[PB Info 2023-Sep-13 17:21:43] || End Time: Wed Sep 13 17:21:43 2023 ||
[PB Info 2023-Sep-13 17:21:43] || Total Time: 1 minute 14 seconds ||
[PB Info 2023-Sep-13 17:21:43] ------------------------------------------------------------------------------
[PB Info 2023-Sep-13 17:21:43] ------------------------------------------------------------------------------
[PB Info 2023-Sep-13 17:21:43] || Parabricks accelerated Genomics Pipeline ||
[PB Info 2023-Sep-13 17:21:43] || Version 4.1.0-1 ||
[PB Info 2023-Sep-13 17:21:43] || Sorting Phase-II ||
[PB Info 2023-Sep-13 17:21:43] ------------------------------------------------------------------------------
[PB Info 2023-Sep-13 17:21:43] progressMeter - Percentage
[PB Info 2023-Sep-13 17:21:43] 0.0 0.00 GB
[PB Info 2023-Sep-13 17:21:48] Sorting and Marking: 5.000 seconds
[PB Info 2023-Sep-13 17:21:48] ------------------------------------------------------------------------------
[PB Info 2023-Sep-13 17:21:48] || Program: Sorting Phase-II ||
[PB Info 2023-Sep-13 17:21:48] || Version: 4.1.0-1 ||
[PB Info 2023-Sep-13 17:21:48] || Start Time: Wed Sep 13 17:21:43 2023 ||
[PB Info 2023-Sep-13 17:21:48] || End Time: Wed Sep 13 17:21:48 2023 ||
[PB Info 2023-Sep-13 17:21:48] || Total Time: 5 seconds ||
[PB Info 2023-Sep-13 17:21:48] ------------------------------------------------------------------------------
[PB Info 2023-Sep-13 17:21:49] ------------------------------------------------------------------------------
[PB Info 2023-Sep-13 17:21:49] || Parabricks accelerated Genomics Pipeline ||
[PB Info 2023-Sep-13 17:21:49] || Version 4.1.0-1 ||
[PB Info 2023-Sep-13 17:21:49] || Marking Duplicates, BQSR ||
[PB Info 2023-Sep-13 17:21:49] ------------------------------------------------------------------------------
[PB Info 2023-Sep-13 17:21:49] BQSR using CUDA device(s): { 0 }
[PB Info 2023-Sep-13 17:21:49] Using PBBinBamFile for BAM writing
[PB Info 2023-Sep-13 17:21:49] progressMeter - Percentage
[PB Info 2023-Sep-13 17:21:59] 0.0 4.49 GB
[PB Info 2023-Sep-13 17:22:09] 0.0 4.49 GB
[PB Info 2023-Sep-13 17:22:19] 0.0 4.49 GB
[PB Info 2023-Sep-13 17:22:29] 0.0 4.49 GB
[PB Info 2023-Sep-13 17:22:39] 88.7 1.81 GB
[PB Info 2023-Sep-13 17:22:49] 100.0 0.00 GB
[PB Info 2023-Sep-13 17:22:49] BQSR and writing final BAM: 60.028 seconds
[PB Info 2023-Sep-13 17:22:49] ------------------------------------------------------------------------------
[PB Info 2023-Sep-13 17:22:49] || Program: Marking Duplicates, BQSR ||
[PB Info 2023-Sep-13 17:22:49] || Version: 4.1.0-1 ||
[PB Info 2023-Sep-13 17:22:49] || Start Time: Wed Sep 13 17:21:49 2023 ||
[PB Info 2023-Sep-13 17:22:49] || End Time: Wed Sep 13 17:22:49 2023 ||
[PB Info 2023-Sep-13 17:22:49] || Total Time: 1 minute 0 seconds ||
[PB Info 2023-Sep-13 17:22:49] ------------------------------------------------------------------------------
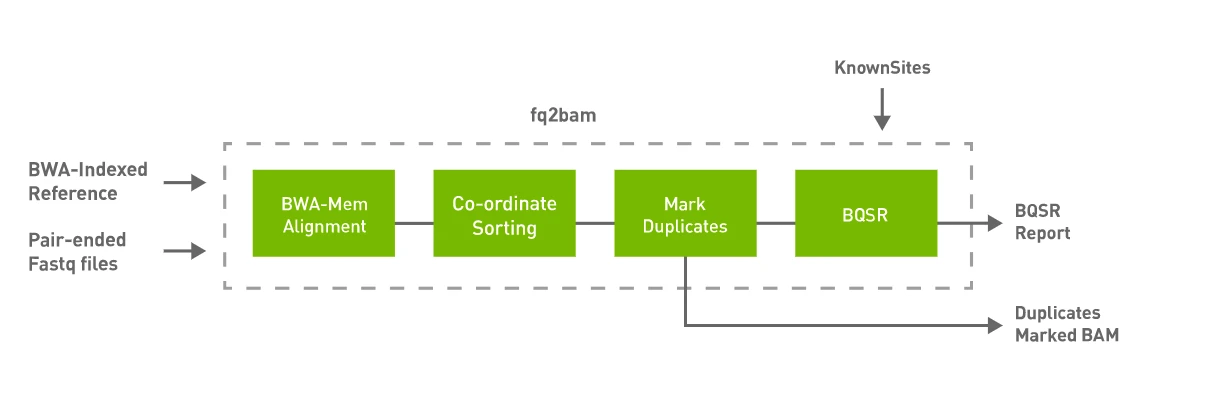
fq2bam performs the following steps.
Compatible CPU-based BWA-MEM, GATK4 Commands
srun -p batch -c 32 --mem=100g --pty bash
singularity run /pfss/containers/gatk.4.4.0.0.sif bash
cd $SCRATCH/parabricks
cd bwa
./bwa mem -t 32 -K 10000000 -R '@RG\tID:SRR9932168.1.1 \tLB:lib1\tPL:bar\tSM:sample\tPU:SRR9932168.1.1 ' \
../human_g1k_v37.fasta \
../SRR9932168_1.fastq \
../SRR9932168_2.fastq | \
gatk SortSam \
--java-options -Xmx30g \
--MAX_RECORDS_IN_RAM 5000000 \
-I /dev/stdin \
-O ../cpu.bam \
--SORT_ORDER coordinate
# for max spot id 5000000 spent 2.72 mins for sorting, 2.2 mins for convert to BAM
[main] CMD: ./bwa mem -t 32 -K 10000000 -R @RG\tID:SRR9932168.1.1 \tLB:lib1\tPL:bar\tSM:sample\tPU:SRR9932168.1.1 /pfss/scratch02/appcara/parabricks/parabricks_sample/Ref/human_g1k_v37.fasta /pfss/scratch02/appcara/parabricks/sratoolkit.3.0.6-centos_linux64/SRR9932168_1.fastq /pfss/scratch02/appcara/parabricks/sratoolkit.3.0.6-centos_linux64/SRR9932168_2.fastq
[main] Real time: 151.130 sec; CPU: 3594.553 sec
INFO 2023-08-01 05:58:33 SortSam Finished reading inputs, merging and writing to output now.
INFO 2023-08-01 05:58:47 SortSam Wrote 10,000,000 records from a sorting collection. Elapsed time: 00:02:43s. Time for last 10,000,000: 14s. Last read position: */*
[Tue Aug 01 05:58:47 GMT 2023] picard.sam.SortSam done. Elapsed time: 2.72 minutes.
Runtime.totalMemory()=1409286144
Tool returned:
0
# generate .dict file
cd $SCRATCH/parabricks
gatk CreateSequenceDictionary -R human_g1k_v37.fasta
# Mark duplicates.
gatk MarkDuplicates \
--java-options -Xmx30g \
-I ./cpu.bam \
-O ./mark_dups_cpu.bam \
-M metrics.txt
# spend 1 mins
# Generate a BQSR report.
gatk IndexFeatureFile -I ./00-common_all.vcf
gatk BaseRecalibrator \
--java-options -Xmx30g \
--input ./mark_dups_cpu.bam \
--output ./recal_cpu.txt \
--known-sites ./00-common_all.vcf \
--reference ./human_g1k_v37.fasta
# spend 1.68 mins
| sample size (bp) | 1M | 5M | 110M |
| 32 core 100g mem | 1.63 mins | 7.56 mins | 140.47 mins |
| 16 core 128g mem A100:1 | 1.66 mins | 2. 33mins | 24.4 mins |
One of the main advantages of Parabricks is its speed. The software can analyze large datasets in a fraction of the time it would take traditional tools to complete the same task. Parabricks is also highly scalable and can analyze datasets of varying sizes without sacrificing performance. It is ideal for researchers and scientists who need to process large amounts of genomic data quickly and efficiently.
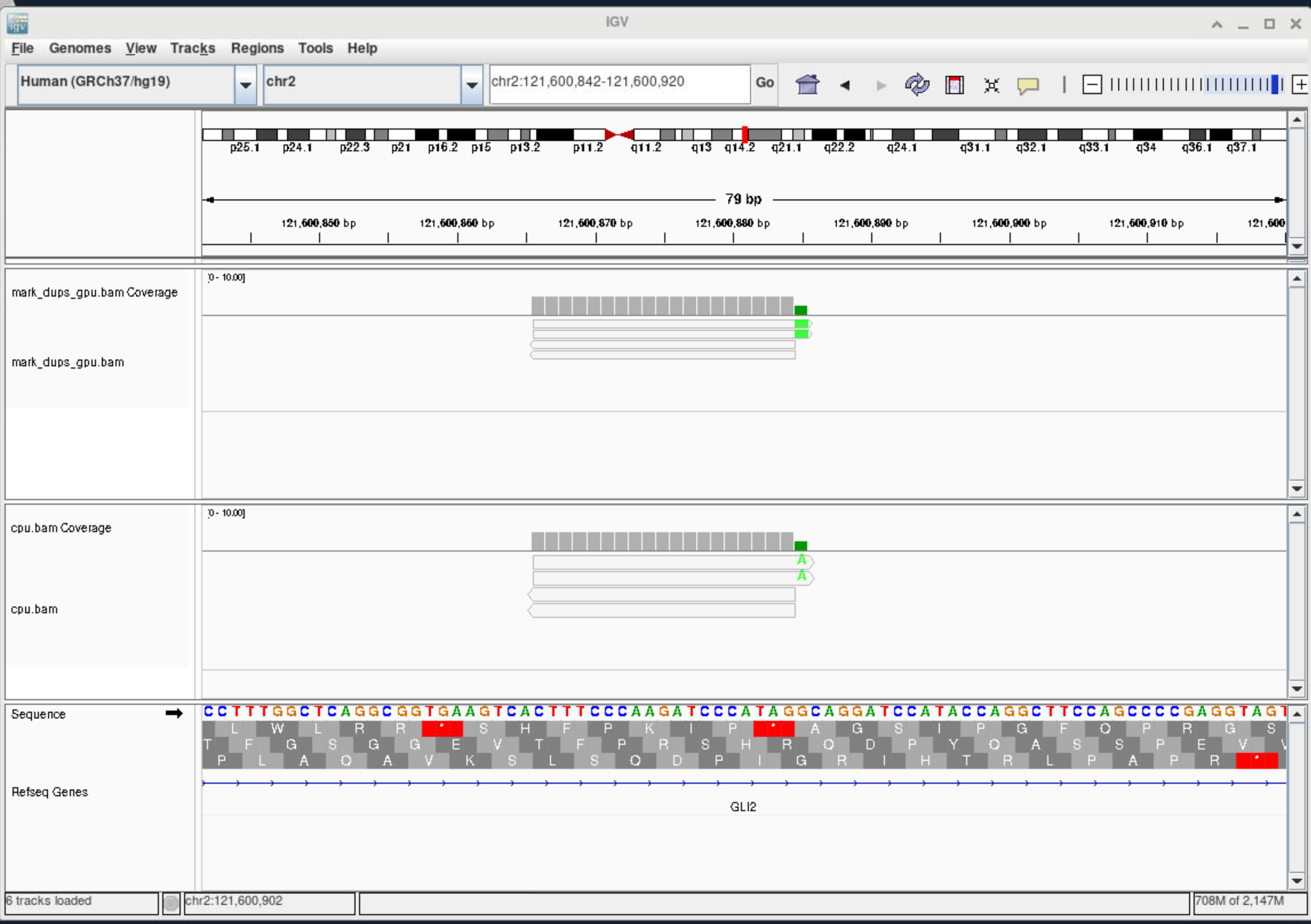
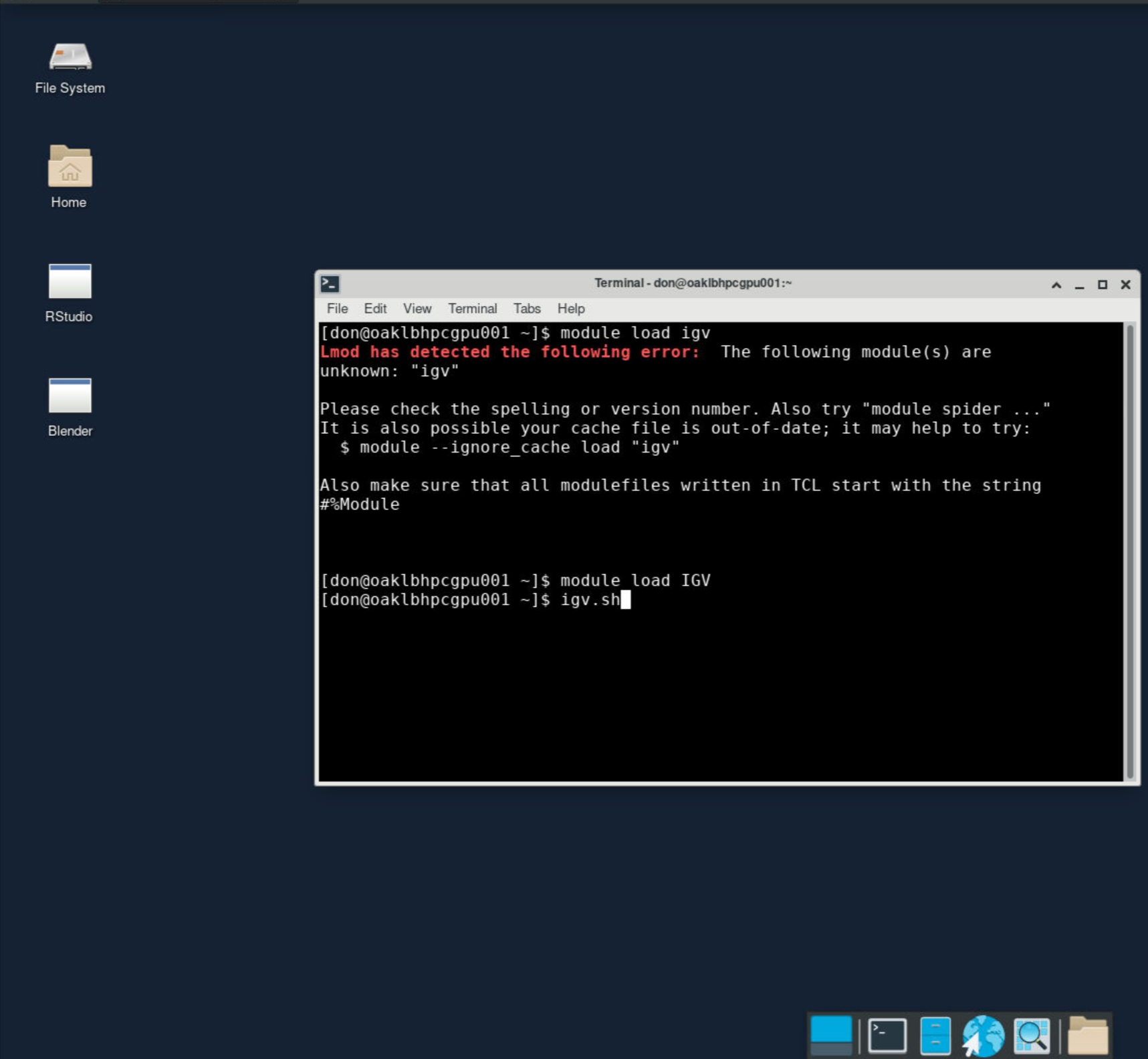
View bam file in IGV
First, start a VNC container, and load module IGV in terminal, after that you can use igv.sh to run IGV
You can use igvtools to index the bam file